1. Electronic Displacements in Organic Molecules
(a) Inductive effect: Inductive effect may be defined as the permanent displacement of electrons forming a
covalent bond towards the more electronegative element or group.
The inductive effect is represented by the symbol, the arrow pointing towards the more electronegative
element or group of elements. Thus in case of n-butyl chloride inductive effect may be represented as below.
Any atom or group, if attracts electrons more strongly than hydrogen, it is said to have a -I effect (electronattracting
or electron-withdrawing), viz NO2, Cl, Br, I, F, COOH OCH3, etc. while if atom or group attracts
electrons less stronlgy than hydrogen it is said to have +I effect (electron repelling or electron-releasing) viz,
CH3, C2H5, Me2CH and Me3C groups. The important atoms or groups which cause negative or positive
inductive effect are arranged below in the order of decreasing effect.
-I (Electron-attracting) groups:
+I (Electron-attracting) groups:
(b) Resonance and Resonance (Mesomeric) effect.
Resonance: The phenomenon in which two or more structures can be written for the true structure of a
molecules, but none of them can be said to represent if uniquely, is referred to as resonacne or mesomerism.
The true structure of the molecule is said to be a resonance hybrid of the various possible alternative
structures which themselves are known as resonating structures or canonical structures. Every two adjacent
resonating structures are represented by inserting a double headed arrow between them. Thus the actual
structure of benzene may be represented in the following two ways.
Necessary conditions for resonance
1. All resonating structures must have the same arrangement of atomic nuclei.
2. The resonating structurs must have the same number of paird and unpaired electrons. However, they differ
in the way of distribution of electrons.
3. The energies of the various limiting structures must be sane or nearly the same.
4. Resonating structures must be planar.
All the resonating structures do not contribute equal to the real molecule and hence only the major
contributing forms are used while represeenting a resonance hybrid.
The mesomeric effect may be defined as the permanent effect in which p electrons are transfered from a
multiple bond to an atom, or from a multiple bond to a single covalent bond or lone pair(s) of p-electrons
from an atom to the adjacent single covalent bond.
Like inductive effect, the mesomeric effect (denoted by M) may be +M and -M. It is +M when the
transference of electron pair is away from the atom and -M when transference of electron pair is toward the
atom. In general.
Some common atoms or groups which cause +M and -M effect are given below
+M groups. -Cl, -Br, -I, -NH2, -NR2, -OH, -OCH3
- M groups. -NO2, -CN, >C=O
(c) Electromeric effect: This type of temporary displacement of electrons take place in compounds
containing multiple covalent bonds (e.g. C=C, C=O, C°N, etc.) or an atom with a lone pair of electrons
adjacent to a covalent bond. The effect involves complete transference of a pair of electrons from a multiple
bond to an atom, or from a multiple bond to another bond, or from an atom with a free pair of electrons to a
bond. it is the p-electrons of a multiple bond, or the p-electrons of an atom, which are transfered. Since the
effect involes complete transference of electrons, it leads to the development of full + and - charges within
the molecule. It is important to note that the electromeric effect is purely a temporary effect and is brought
into play only the requirement of attacking reagent, it vanishes out as soon as the attacking reagent is
removed from reaction mixture.
for more details please visit: http://www.gurukpo.com/
(a) Inductive effect: Inductive effect may be defined as the permanent displacement of electrons forming a
covalent bond towards the more electronegative element or group.
The inductive effect is represented by the symbol, the arrow pointing towards the more electronegative
element or group of elements. Thus in case of n-butyl chloride inductive effect may be represented as below.
Any atom or group, if attracts electrons more strongly than hydrogen, it is said to have a -I effect (electronattracting
or electron-withdrawing), viz NO2, Cl, Br, I, F, COOH OCH3, etc. while if atom or group attracts
electrons less stronlgy than hydrogen it is said to have +I effect (electron repelling or electron-releasing) viz,
CH3, C2H5, Me2CH and Me3C groups. The important atoms or groups which cause negative or positive
inductive effect are arranged below in the order of decreasing effect.
-I (Electron-attracting) groups:
+I (Electron-attracting) groups:
(b) Resonance and Resonance (Mesomeric) effect.
Resonance: The phenomenon in which two or more structures can be written for the true structure of a
molecules, but none of them can be said to represent if uniquely, is referred to as resonacne or mesomerism.
The true structure of the molecule is said to be a resonance hybrid of the various possible alternative
structures which themselves are known as resonating structures or canonical structures. Every two adjacent
resonating structures are represented by inserting a double headed arrow between them. Thus the actual
structure of benzene may be represented in the following two ways.
Necessary conditions for resonance
1. All resonating structures must have the same arrangement of atomic nuclei.
2. The resonating structurs must have the same number of paird and unpaired electrons. However, they differ
in the way of distribution of electrons.
3. The energies of the various limiting structures must be sane or nearly the same.
4. Resonating structures must be planar.
All the resonating structures do not contribute equal to the real molecule and hence only the major
contributing forms are used while represeenting a resonance hybrid.
The mesomeric effect may be defined as the permanent effect in which p electrons are transfered from a
multiple bond to an atom, or from a multiple bond to a single covalent bond or lone pair(s) of p-electrons
from an atom to the adjacent single covalent bond.
Like inductive effect, the mesomeric effect (denoted by M) may be +M and -M. It is +M when the
transference of electron pair is away from the atom and -M when transference of electron pair is toward the
atom. In general.
Some common atoms or groups which cause +M and -M effect are given below
+M groups. -Cl, -Br, -I, -NH2, -NR2, -OH, -OCH3
- M groups. -NO2, -CN, >C=O
(c) Electromeric effect: This type of temporary displacement of electrons take place in compounds
containing multiple covalent bonds (e.g. C=C, C=O, C°N, etc.) or an atom with a lone pair of electrons
adjacent to a covalent bond. The effect involves complete transference of a pair of electrons from a multiple
bond to an atom, or from a multiple bond to another bond, or from an atom with a free pair of electrons to a
bond. it is the p-electrons of a multiple bond, or the p-electrons of an atom, which are transfered. Since the
effect involes complete transference of electrons, it leads to the development of full + and - charges within
the molecule. It is important to note that the electromeric effect is purely a temporary effect and is brought
into play only the requirement of attacking reagent, it vanishes out as soon as the attacking reagent is
removed from reaction mixture.
for more details please visit: http://www.gurukpo.com/


 ).
).
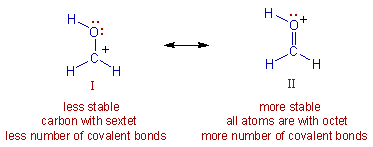
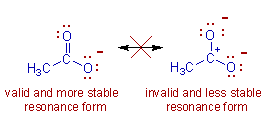
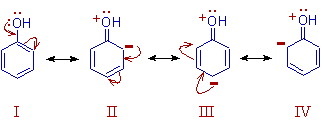
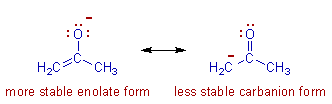
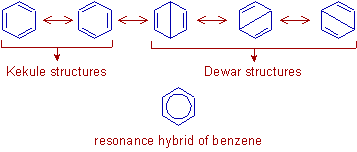







 Figure 1: Newman projection of ethane
Figure 1: Newman projection of ethane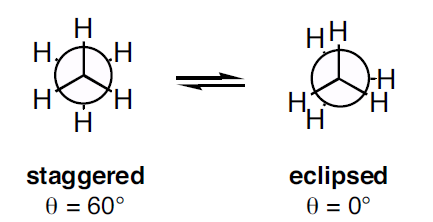 Figure 2: Conformations of ethane - staggered and eclipsed
Figure 2: Conformations of ethane - staggered and eclipsed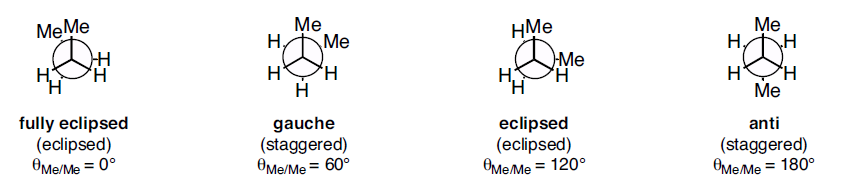 Figure 3: Conformations of butane: fully eclipsed, gauche, eclipsed, and anti
Figure 3: Conformations of butane: fully eclipsed, gauche, eclipsed, and anti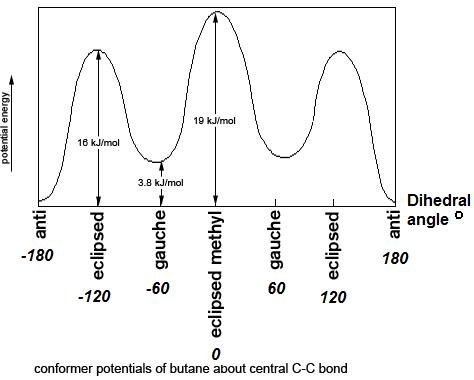 Figure 4: Energy diagram for conformations of butane as a function of dihedral angle
Figure 4: Energy diagram for conformations of butane as a function of dihedral angle












{kind=link}